
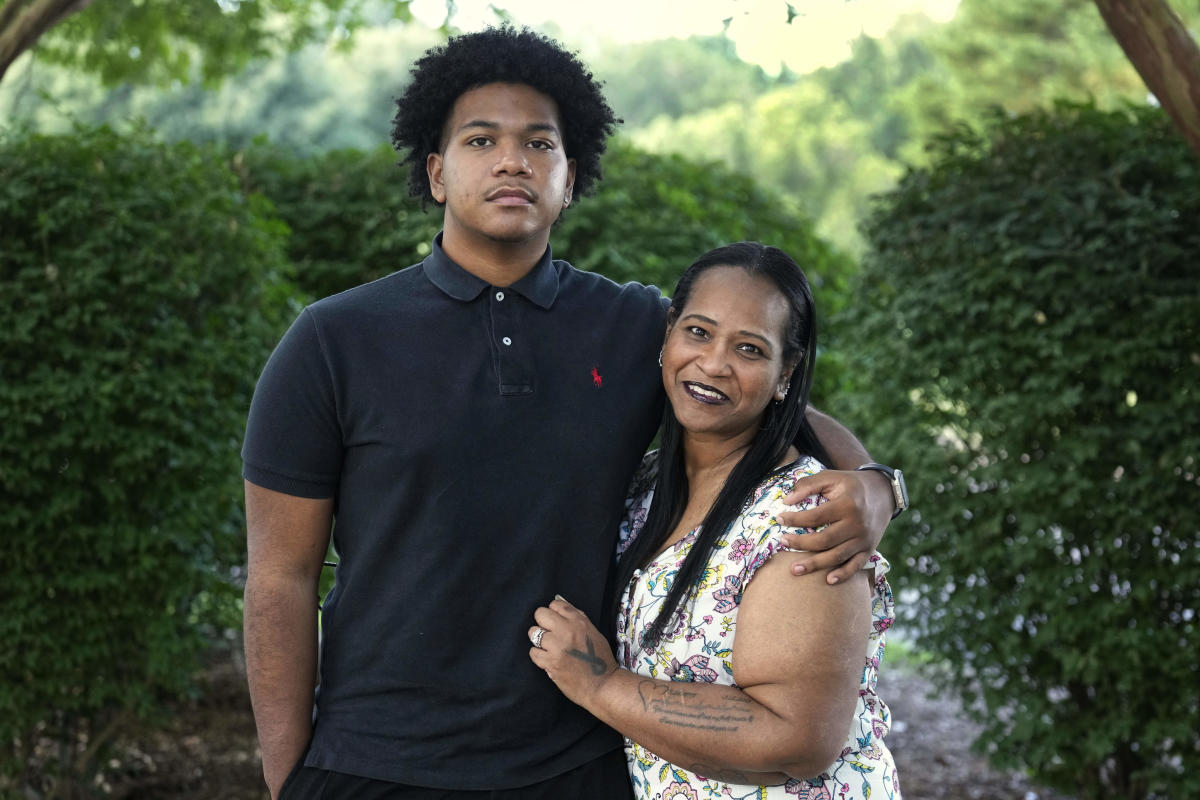
تواجه “روبن ألدرمان” واقعًا مؤلمًا: العلاج الجيني قد يعالج نقص المناعة الوراثي النادر الذي يعاني منه ابنها “كامدن”. لكنها غير متاحة له.
في عام 2022، توقفت شركة Orchard Therapeutics ومقرها لندن عن الاستثمار في علاج تجريبي لحالة متلازمة ويسكوت ألدريتش. ولا توجد دراسات للعلاج الجيني يمكنه الانضمام إليها.
قالت ألدرمان، التي تدافع عن ابنها البالغ من العمر 21 عاماً منذ أن كان طفلاً: “نشعر وكأننا المنسيين”.
ويعاني نحو 350 مليون شخص حول العالم من أمراض نادرة، معظمها وراثية. لكن كل اضطراب من الاضطرابات الفردية البالغ عددها 7000 يؤثر ربما على عدد قليل من كل مليون شخص أو أقل. لا يوجد حافز تجاري كبير لتطوير هذه العلاجات التي تستخدم لمرة واحدة أو تسويقها لإصلاح الجينات المعيبة أو استبدالها بجينات سليمة. وهذا يترك عائلات مثل آلدرمان تتدافع للحصول على المساعدة ويحاول البعض جمع الأموال بأنفسهم من أجل علاجات قد لا تأتي أبدًا.
وقال الدكتور جوليو كوسو، أستاذ الطب التجديدي في جامعة مانشستر في إنجلترا: “كان هؤلاء الأطفال مؤسفين مرتين: أولا، لأنهم أصيبوا بمرض وراثي، وثانيا، لأن المرض نادر للغاية لدرجة أنه لا أحد يهتم”. “الشركات تريد تحقيق الربح.”
يقول العلماء إن هذه الديناميكية تهدد بإحباط التقدم في مجال العلاج الجيني الناشئ، مما يؤدي إلى محو إمكانات نوع جديد من الطب، تمامًا كما يشير دفق مستمر من الأبحاث نحو علاجات واعدة لمختلف الاضطرابات. يبحث الباحثون عن حلول، وغالبًا ما يلجأون إلى المنظمات الخيرية ومجموعات المرضى والحكومات.
أعلنت مؤسسة خيرية إيطالية كبرى في فبراير/شباط أنها ستتولى معالجة علاج ويسكوت-ألدريتش الذي كان أوركارد يسعى إليه. وساعدت إحدى مؤسسات مؤسسة مكافحة العمى الخيرية في إطلاق شركة، Opus Genetics، لتعزيز أعمال العلاج الجيني التي قام بها الباحث في جامعة بنسلفانيا الدكتور جان بينيت وزميله.
ومن نواحٍ عديدة، كان هذا الجهد مستوحى من عائلات المرضى.
“بعضهم لديه مبيعات خبز. قال بينيت: “قامت إحدى العائلات برهن منزلها للتبرع ببعض المال لإجراء دراسة عن مرضهم النادر”. “أنا فقط أشعر بالمسؤولية لمساعدتهم.”
آلام العائلات
لقد واجه آلدرمان سنوات من الألم والإحباط.
تم تشخيص حالة كامدن ألدرمان عندما كان طفلاً مصابًا بمرض ويسكوت ألدريتش، الناجم عن طفرة جينية على الكروموسوم X. وهو يؤثر في المقام الأول على الأولاد – ما يصل إلى 10 من كل مليون – ويمكن أن يسبب التهابات متكررة، والأكزيما، والنزيف المفرط.
عندما كان طفلاً صغيرًا، قام الأطباء بإزالة طحاله بسبب النزيف غير المنضبط. عندما كان صبيًا صغيرًا، انتهى به الأمر في المستشفى عدة مرات وقيل له إنه لا يستطيع لعب البيسبول.
أحد العلاجات هو زرع نخاع العظم. لكنه أسود وله تراث كوري، مما يجعل من الصعب العثور على متبرع – فمن المرجح أن يتطابق الأشخاص مع شخص من أسلاف أو خلفيات عرقية مماثلة. يتذكر روبن ألدرمان أن أحد الأطباء قال: “في الأساس، الفرصة الوحيدة أمام ابنك للشفاء ستكون العلاج الجيني”.
وأخبرها أيضًا أن الباحثين لم يقبلوا بعد ذلك المقيمين في الولايات المتحدة في تجربة سريرية، الأمر الذي “لقد حطم قلبي نوعًا ما”، على حد قولها.
اليوم، كامدن ألدرمان هو طالب صاعد في جامعة ولاية كارولينا الشمالية الزراعية والتقنية. وهو يتناول البنسلين يوميًا ويحقن نفسه أسبوعيًا بالجلوبيولين المناعي تحت جلده، مما يساعد على مكافحة العدوى. ومع ذلك، فقد دخل المستشفى عدة مرات في السنوات الأخيرة وأصيب بمشكلة في الكلى.
ورغم أنه لا ينظر إلى العلاج الجيني باعتباره علاجًا شاملاً، إلا أنه قال: “إنه سيساعدني على عيش حياة أسهل نوعًا ما”.
وقد ثبت أن هذا صحيح بالنسبة للمرضى الذين خضعوا للعلاج التجريبي، مثل ابن الدكتورة بريا ستيفن البالغ من العمر 14 عامًا، والذي شارك في تجربة سريرية في إيطاليا قبلت الأمريكيين في ذلك الوقت.
بينما تشعر ستيفن بالامتنان، قالت إنها لا تستطيع منع نفسها من الشعور بالذنب لأن عائلتها حصلت على فرصة لم يحصل عليها الآخرون: “من غير المقبول أخلاقياً أن يكون لدينا علاج نعلم أنه فعال، ونعلم أنه آمن، وأن الناس جميعًا فجأة لا يمكن الوصول.”
لفترة من الوقت، بدا أن العلاج الجيني لـ Wiskott-Aldrich كان في طريقه للتوفر على نطاق أوسع. وقال فريديريك ريفا، الرئيس التنفيذي لشركة Genethon، وهي منظمة بحثية فرنسية غير ربحية، إنها قامت برعاية تجارب سريرية واعدة ولكن لم يكن لديها التمويل لمواصلة التطوير.
نقلت شركة الأدوية جلاكسو سميث كلاين علاجًا آخر إلى شركة أوركارد، التي أعلنت في عام 2019 أنها حصلت على تصنيف من إدارة الغذاء والدواء الأمريكية بهدف تسريع التطوير والمراجعة. لكن شركة Orchard أوقفت الاستثمار في هذا العلاج وعلاجين آخرين للأمراض النادرة منذ عامين، حيث قال الرئيس التنفيذي الدكتور بوبي جاسبار إن الشركة تتعاطف مع العائلات المتضررة وستبحث عن طرق أخرى لتطوير العلاجات.
وقال ريفا: “هناك عدد كبير من الأمراض التي يمكن أن تستفيد من العلاج الجيني ولكن لا يوجد نموذج ربحية لها لأن الاستثمار في الأبحاث مرتفع، وتكلفة الإنتاج مرتفعة، وعدد المرضى منخفض للغاية”.
معظم الحالات الوراثية نادرة، حيث يؤثر كل منها على أقل من 200 ألف شخص في الولايات المتحدة في أي وقت. لم تتجاوز الأبحاث المراحل المبكرة بالنسبة للعديد منهم.
تعاني ابنة لاسي هندرسون، إستيلا، البالغة من العمر 5 سنوات، من شلل نصفي متناوب في مرحلة الطفولة، وهي حالة عصبية تؤثر على 300 شخص في الولايات المتحدة. وتعاني إستيلا من تأخر إدراكي، ولديها قدرة محدودة على استخدام يديها، وتصاب بالشلل المؤقت في جزء أو كل جسدها. قال هندرسون. يمكن للأدوية أن تحد من الأعراض، لكن لا يوجد علاج.
تقوم عائلتها في ولاية أيوا بجمع التبرعات من خلال GoFundMe وموقع ويب لتطوير العلاج الجيني. لقد جلبوا حوالي 200 ألف دولار.
قال هندرسون: “لدينا ثلاثة مشاريع مختلفة مع باحثين مختلفين. لكن المشكلة هي أن كل شيء يعاني من نقص التمويل”.
حوافز “منحرفة”.
يقول العلماء إن المثبطات المالية تعصف بهذه العملية، بدءًا من اكتشاف الأدوية وحتى تطويرها.
وقال الدكتور دونالد كون، أستاذ علم الأحياء الدقيقة والمناعة وعلم الوراثة الجزيئية في جامعة كاليفورنيا، لوس أنجلوس، إن حجم العمل المطلوب من المختبر إلى الاختبارات البشرية ومن خلال عملية الموافقة على الأدوية “مكلف بشكل لا يصدق”.
وقال إنه في العامين الماضيين، جفت الاستثمارات في العلاج الجيني إلى حد كبير.
وقال كوهن: “إذا كان عليك إنفاق 20 مليون دولار أو 30 مليون دولار للحصول على الموافقة وكان لديك خمسة أو 10 مرضى سنويًا، فمن الصعب الحصول على عائد على الاستثمار”. “لذلك لدينا علاجات ناجحة وآمنة، ولكن العناصر المالية والاقتصادية هي التي تحد من أن تصبح أدوية معتمدة.”
وقال فرانسوا فيجنولت، الرئيس التنفيذي لشركة سياتل للتكنولوجيا الحيوية شيب ثيرابيوتيكس، إن معظم شركات التكنولوجيا الحيوية تصبح عامة في نهاية المطاف، ويجب أن تركز على أرباح المساهمين.
«إن مجلس الإدارة هو الشيء الذي يعيق الطريق؛ وقال فيجنولت، الذي تعد شركته مملوكة للقطاع الخاص: “إنهم يحاولون تحقيق أقصى قدر من المكاسب”. “هذا مجرد جشع. هذا مجرد حافز غير متوازن بين هيكل الشركة وما يجب أن نفعله وهو أمر جيد للعالم.
وحتى عندما تصل العلاجات إلى الأسواق، فإنها قد لا تبقى هناك. في نفس العام الذي توقفت فيه شركة Orchard عن الاستثمار في علاج Wiskott-Aldrich، توقفت أيضًا عن توزيع دواء يسمى Strimvelis، المعتمد في أوروبا لعلاج المرض النادر ADA-SCID، أو “متلازمة الصبي الفقاعي”.
“تحدي هائل”
كلير بوث، أستاذة العلاج الجيني وعلم مناعة الأطفال في جامعة كوليدج لندن، هي من بين أولئك الذين يعملون من أجل التغيير. شاركت في تأسيس برنامج الوصول إلى العلاجات الجينية للأمراض النادرة، والذي يجمع أشخاصًا من جميع أنحاء أوروبا يمثلون المجموعات الأكاديمية والمدافعين عن المرضى والمنظمين والممولين وصانعي الأدوية. إنهم يأملون في إنشاء منظمة غير ربحية مستقلة يمكنها دعم ترخيص السوق والوصول إلى العلاجات غير المستدامة تجاريًا.
وهناك جهد ذو صلة في الولايات المتحدة، وهو اتحاد العلاج الجيني المصمم حسب الطلب، والذي تم تنظيمه من قبل مؤسسة المعاهد الوطنية للصحة ويضم إدارة الغذاء والدواء، والعديد من معاهد المعاهد الوطنية للصحة، والعديد من شركات الأدوية والمنظمات غير الربحية. تتضمن أهداف المجموعة دعم مجموعة من التجارب السريرية واستكشاف طرق لتبسيط العمليات التنظيمية.
ويحاول بعض الباحثين معالجة المشكلة علميا. قالت الدكتورة آنا جريكا إن معهد برود التابع لمعهد ماساتشوستس للتكنولوجيا وجامعة هارفارد قد أطلقا جهدًا للنظر في القواسم المشتركة وراء الظروف المختلفة – أو العقد، والتي يمكن تشبيهها بالفروع التي تجتمع في جذع شجرة. إن إصلاح العقد باستخدام علاجات جينية أو علاجات أخرى، بدلا من “أخطاء إملائية” معينة في الحمض النووي المسؤول عن اضطراب واحد، يمكن أن يعالج أمراض متعددة في وقت واحد.
قال غريكا، عضو برود: “ما يفعله هذا هو أنه يزيد عدد المرضى الذين يمكنهم الاستفادة من العلاج”. “كما أنه يجعل الأمر أسهل أو أكثر جاذبية لأي شخص، مثل شركة الأدوية الحيوية، للمضي قدمًا بالمشروع ومحاولة تقديمه إلى العيادات، لأنه سيكون لديهم سوق أكبر.”
وفي الوقت نفسه، تتعاون الأسر المتضررة مع بعضها البعض ومع العلماء للمساعدة في تحريك الإبرة. تم إنشاء جينثون من قبل جمعية من المرضى وأقاربهم لتطوير علاجات للعديد من الأمراض النادرة. وزعيم المؤسسة المشاركة في Opus Genetics لديه طفل مصاب بمرض شبكي وراثي نادر.
هناك أيضًا أمل جديد للعائلات التي تتعامل مع مرض ويسكوت ألدريتش ومرض الفقاعة. في العام الماضي، تولت مؤسسة Telethon في إيطاليا مسؤولية إنتاج وتوزيع Strimvelis. أعلنت المؤسسة الخيرية هذا العام أنه تم اختيارها لبرنامج تجريبي لوكالة الأدوية الأوروبية، والذي يمكن أن يساعد في توجيه العلاج الجيني لـ Wiskott-Aldrich من خلال العملية التنظيمية هناك.
ومع ذلك، يقول العلماء إن هذه الجهود لا تنفي المأزق المالي الأكبر المحيط بعلاجات الأمراض النادرة، وقد يستغرق الأمر بعض الوقت قبل أن تصبح هذه العلاجات الجينية متاحة للمرضى في جميع أنحاء العالم.
وقال بوث: “هذا تحدٍ هائل، ولست متأكداً تماماً من أننا سنكون قادرين على التغلب عليه”. “لكن علينا أن نجربها لأننا أمضينا عقودًا وملايين في صنع هذه العلاجات التحويلية. وإذا لم نحاول، فسيبدو الأمر وكأنه نهاية حقبة”.
____
يتلقى قسم الصحة والعلوم في وكالة أسوشيتد برس الدعم من مجموعة الإعلام العلمي والتعليمي التابعة لمعهد هوارد هيوز الطبي. AP هي المسؤولة الوحيدة عن جميع المحتويات.