
ملحوظة المحرر: إليزابيث يوكو، حاصلة على دكتوراه، عالمة في الأخلاقيات الحيوية وكاتبة ظهرت أعمالها في صحيفة نيويورك تايمز، ورولينج ستون، وواشنطن بوست، وذا أتلانتيك وأماكن أخرى. وهي أيضًا أستاذة مساعدة في جامعة فوردهام. الآراء المعبر عنها هنا هي آراءها. يقرأ مزيد من الرأي على سي إن إن.
في ديسمبر/كانون الأول، وافقت إدارة الغذاء والدواء الأمريكية (FDA) على أول علاجين جينيين قائمين على الخلايا لعلاج مرض فقر الدم المنجلي (SCD) لدى الأشخاص الذين تبلغ أعمارهم 12 عامًا فما فوق: Casgevy وLyfgenia. منذ ذلك الحين، تزايدت الأسئلة والمخاوف حول تكلفة هذه العلاجات وإمكانية الوصول إليها – بما في ذلك من وزير الصحة والخدمات الإنسانية كزافييه بيسيرا، الذي سلط الضوء باستمرار على الحواجز المالية التي تقف بين الأشخاص الذين يعيشون مع مرض فقر الدم المنجلي و هذه العلاجات الجينية الجديدة.
في حين أن كل من Casgevy وLyfgenia يعتبران إنجازين خارقين، فإن Casgevy مهم لسبب آخر: إنه أول استخدام سريري معتمد من إدارة الغذاء والدواء الأمريكية لتحرير الجينات CRISPR-Cas9 (يشار إليه عادةً باسم CRISPR) لعلاج أي حالة. (رغم أن ليفجينيا تعتمد أيضا على تعديل الجينات، فإنها تستخدم تكنولوجيا مختلفة وأكثر رسوخا).
يشير تحرير الجينات إلى طرق مختلفة يمكن استخدامها لإدخال تسلسل الحمض النووي أو استبداله أو إزالته في جينومات الكائنات الحية، بما في ذلك البشر. تعمل تقنية كريسبر مثل المقص الجيني لقص الحمض النووي ثم تحريره في مكان معين، وقد اكتسبت استحسانًا بين الباحثين لكونها أسهل وأسرع وأكثر دقة وأقل تكلفة من التقنيات السابقة.
يعد هذا التطبيق العلاجي الأول لكريسبر الذي وافقت عليه إدارة الغذاء والدواء الأمريكية، بلا شك، خطوة مهمة إلى الأمام في مجال العلاج الجيني وممارسة الطب. وكما هو الحال مع أي تكنولوجيا طبية ناشئة، قوبل الاستخدام الأولي لأداة تحرير الجينات المبتكرة بمزيج من الترقب والقلق.
من المؤكد أن هناك قضايا أخلاقية مؤثرة ــ وأبرزها المسائل المتعلقة بإمكانية الوصول إلى العلاجات الجديدة التي تبلغ قيمتها ملايين الدولارات. ولكن نظرًا لأن أداة تحرير الجينات أصبحت جزءًا من مجموعة الأدوات لعلاج عدد متزايد من الحالات، فمن المهم عدم الخلط أو حتى ربط تطبيق كريسبر هذا مع الاستخدامات الأكثر إشكالية للتكنولوجيا.
لقد طال انتظار Casgevy وLyfgenia
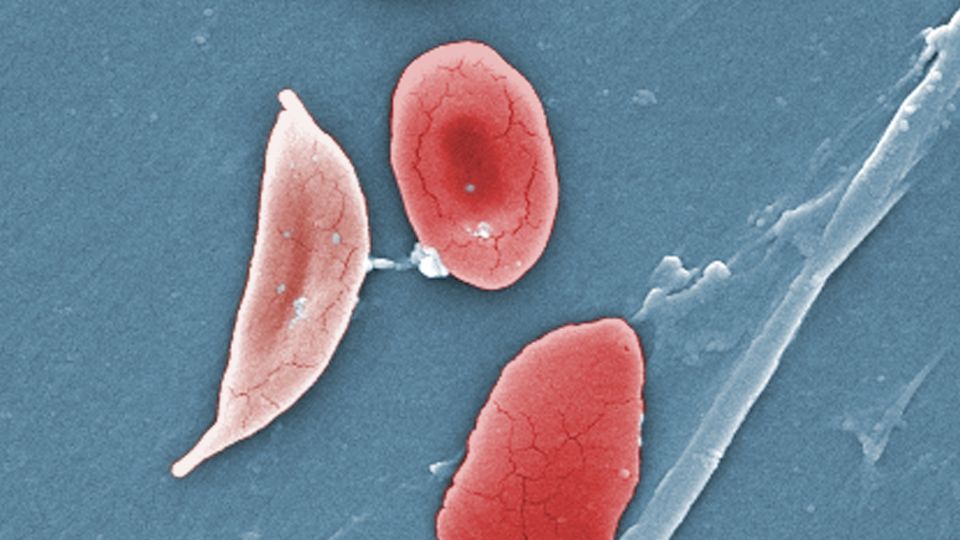
تتميز مجموعة اضطرابات خلايا الدم الحمراء الموروثة والمعروفة مجتمعة باسم SCD بتشوهات تجعل خلايا الدم الحمراء لدى الشخص لزجة وعلى شكل حرف C بدلاً من أن تكون مستديرة. تسبب هذه الخلايا المنجلية انسدادًا في الأوعية الدموية للمريض، مما يؤدي غالبًا إلى الألم ومضاعفات أخرى.
وتقدر المراكز الأمريكية لمكافحة الأمراض والوقاية منها أنها تؤثر على ما يقرب من 100 ألف شخص في الولايات المتحدة. يعد مرض SCD أكثر انتشارًا بين الأشخاص ذوي الأصول الأفريقية، وفقًا للمعهد الوطني للقلب والرئة والدم. تشير دراسة نُشرت في مايو 2023 إلى أنه من بين ما يقرب من 75000 شخص تم إدخالهم إلى المستشفى بسبب مرض فقر الدم المنجلي في الولايات المتحدة بين عامي 2016 و2018، كان 93% منهم من السود. وفي الولايات المتحدة، يُعتقد أن حوالي 20 ألف شخص مصابون بنوع من المرض شديد الخطورة بما يكفي للتأهل لهذه العلاجات القائمة على الجينات.
يعاني حوالي 20% من مرضى SCD في الولايات المتحدة من شكل أكثر خطورة من المرض، يتميز بنوبات مؤلمة ومتكررة، وفقًا لتقديرات الدكتور ديفيد أ. ويليامز، رئيس قسم أمراض الدم/الأورام في مستشفى بوسطن للأطفال وأستاذ طب الأطفال في جامعة بوسطن. كلية الطب بجامعة هارفارد. لم يشارك ويليامز في التجارب السريرية لأي من العلاجات الجينية الجديدة، ولكنه استشار في عملية الموافقة التنظيمية لكلا المصنعين.
قال لي: “إنه مشابه للألم الذي تشعر به بسبب نوبة قلبية – والذي غالبًا ما يعبر عنه الناس بأنه الألم الأكثر إيلامًا الذي شعروا به على الإطلاق”. إن تكرار هذه النوبات المؤلمة وعدم القدرة على التنبؤ بها يؤثر فعليًا على كل جانب من جوانب حياة الشخص، مما قد يجعل من الصعب الذهاب إلى المدرسة باستمرار أو الاستمرار في وظيفة.
ومع ذلك، على الرغم من آثاره المنهكة ــ وحقيقة أن مرض فقر الدم المنجلي أصبح في عام 1949 أول مرض يتم تحديده على المستوى الجزيئي ــ ظلت الأبحاث في مجال العلاج والشفاء تعاني من نقص التمويل لعقود من الزمن. وكان هذا، إلى حد كبير، نتيجة لما وصفته هارييت أ. واشنطن في كتابها “الفصل العنصري الطبي”، بأنه “الاعتقاد الخاطئ” بأن مرض فقر الدم المنجلي “كان حالة عنصرية تصيب الأميركيين من أصل أفريقي فقط”.
في حين أن هناك أربعة أدوية تستخدم لعلاج جوانب مرض فقر الدم المنجلي – تمت الموافقة على ثلاثة منها بين عامي 2017 و 2019 – فإن العلاج المحتمل الوحيد لهذه الحالة هو زرع الخلايا الجذعية.
تتمتع عمليات زرع الأعضاء من متبرعين أشقاء متطابقين بأفضل فرصة للنجاح، ولكن، كما يشير ويليامز، تشير التقديرات إلى أن أقل من 20% من مرضى فقر الدم المنجلي لديهم متبرعون أشقاء مناسبون. على الرغم من أنه من الممكن أيضًا استخدام الخلايا الجذعية من شخص لا تربطه صلة قرابة بالمتلقي، يقول ويليامز إن “عدد المتبرعين المتاحين الذين يتطابقون مع شخص من أصل أمريكي من أصل أفريقي أو من أصل لاتيني منخفض” ولا يلبي الطلب.
أدخل العلاجين المعتمدين حديثًا. على الرغم من أن كلاهما يتضمن عمليات زرع، إلا أنه يتم استخدام الخلايا الجذعية الخاصة بالمريض، مما يلغي الحاجة إلى متبرع والمخاطر المصاحبة له.
بينما يستخدم Casgevy تقنية CRISPR لتحرير الحمض النووي في الخلايا الجذعية لدم المريض بحيث تبدأ في إنتاج الهيموجلوبين الصحي، تستخدم Lyfgenia مكونات من فيروس لتوصيل جين جديد إلى الخلايا الجذعية في دم المريض التي تنتج الهيموجلوبين الصحي.
إن وجود علاجين مبتكرين لعلاج مرض يؤثر على مجموعة سكانية تعاني من نقص الخدمات بشكل مزمن يعد بمثابة أخبار كبيرة وإيجابية إلى حد كبير.
كريسبر يعقد المحادثة
لقد طغى تركيز الليزر على تقنية كريسبر، من نواحٍ عديدة، على تقديم علاجات مرض فقر الدم المنجلي التي طال انتظارها. على الرغم من أن الموافقة الافتتاحية لإدارة الغذاء والدواء الأمريكية على علاج يستخدم أداة تحرير الجينات جديرة بالملاحظة، فإن التركيز على تقنية كريسبر – التي يفهم الكثير من الناس أن لديها على الأقل بعض التطبيقات الإشكالية أخلاقيًا – يمكن أن يلقي بظلاله غير المستحقة على العلاجات الجديدة.
غالبًا ما ينبع الارتباك من حقيقة أنه يمكن استخدام تقنية كريسبر لتعديل فئتين مختلفتين من الخلايا: الخلايا الجرثومية (البويضات والحيوانات المنوية)، والخلايا الجسدية، التي تشكل بقية الجسم، بما في ذلك خلايا الدم التي يغيرها كاسجيفي. إن تحرير الخلايا الجسدية يؤثر فقط على الفرد؛ بل على العكس من ذلك، فإن أي تغييرات تطرأ على المادة الوراثية للأجنة أو الخلايا الجرثومية تنتقل إلى الأجيال اللاحقة.
وبسبب التساؤلات الأخلاقية التي تثار عندما لا تؤثر التعديلات الجينية على المريض فحسب، بل على نسله أيضا، فقد حظي تعديل السلالة الجرثومية، بشكل مفهوم، بالقسم الأكبر من اهتمام عامة الناس عندما يتعلق الأمر بتقنية كريسبر. ولكي نكون واضحين، فإن استخدام “المقص الجيني” لا علاقة له باستخدامه في كاسجيفي.
يقول ويليامز: “هناك القليل من الجدل حول تحرير جينات الخلايا الجسدية: لقد كنا نفعل ذلك الآن لسنوات عديدة”. “وهذا يتناقض إلى حد كبير مع تحرير جينات الخلايا الجرثومية، المحظور.”
ارتفاع تكلفة العلاج
في هذه المرحلة، ليس من الواضح مقدار المبلغ الذي سيتعين على مرضى فقر الدم المنجلي دفعه مقابل العلاج، وما إذا كانت شركات التأمين ستغطي أيًا منهما. ما نعرفه هو أن الشركات المصنعة سوف تبيع منتجات Casgevy وLyfgenia لتجار الجملة في الولايات المتحدة – الذين يبيعون المنتجات إلى الصيدليات والمرافق الطبية بسعر أعلى – مقابل 2.2 مليون دولار و3.1 مليون دولار على التوالي.
أعلنت وزارة الصحة والخدمات الإنسانية الأمريكية مؤخرًا أنها ستقدم نموذج تسعير جديد في عام 2025 مما يجعل Casgevy وLyfgenia في متناول ما يقدر بنحو 50٪ إلى 60٪ من الأشخاص الذين يعيشون مع SCD المسجلين في Medicaid. البيان – الذي يشير أيضًا إلى أن “الاستشفاء والحلقات الصحية الأخرى المرتبطة بـ SCD تكلف النظام الصحي ما يقرب من 3 مليارات دولار سنويًا” – لا يذكر أي شيء عن جعل العلاجات الجديدة ميسورة التكلفة لمرضى SCD غير المسجلين في برنامج Medicaid. (تشير التقارير الأخيرة الصادرة عن مؤسسة KFF إلى أن شركة Blue Cross Blue Shield ستغطي هذه العلاجات، على الرغم من أن التكلفة المباشرة للمرضى قد تظل مرتفعة).
لاحظت شركة Bluebird Bio، الشركة المصنعة لـ Lyfgenia، أن السعر “اعترافًا بالقيمة التي قد يقدمها العلاج من خلال فوائد سريرية قوية ومستدامة” والتأثير الذي يمكن أن يحدثه العلاج على تقليل تكلفة الرعاية الصحية للتعامل مع المرض على مدار العام. مدى حياة المريض. قدرت الشركتان تكلفة إدارة مرض فقر الدم المنجلي بما يتراوح بين 4 ملايين دولار و6 ملايين دولار على مدى عمر المريض الذي يعاني من أزمات الألم المتكررة.
إن التكاليف المرتفعة للغاية للعلاجات الجديدة من شأنها أن تجعلها غير متاحة للعديد من المرضى في الولايات المتحدة، بل وأكثر من ذلك بعيدة عن متناول الأشخاص الذين يعيشون في البلدان المنخفضة والمتوسطة الدخل حيث تكون أمراض القلب والأوعية الدموية أكثر شيوعا.
وهذا أمر غير عادل ليس فقط لأن الأشخاص الذين يعيشون في جزء واحد من العالم لا يستحقون العلاج أكثر من غيرهم، ولكن أيضًا لأنه من المرجح أن يؤدي إلى تفاقم الفوارق الصحية القائمة – مما يحد في الأساس من الإصابة بأمراض القلب التاجية لدى الأشخاص ذوي الدخل المنخفض.
وزن المخاطر والفوائد
مثل أي علاج أو إجراء تمت الموافقة عليه حديثًا، فإن النتائج طويلة المدى لـ Casgevy وLyfgenia – بما في ذلك ما إذا كانا سيكونان علاجًا مدى الحياة لمرض SCD، كما هو متوقع – ليست معروفة بعد. بينما تستخدم ليفجينيا تقنية العلاج الجيني التي تم استخدامها في المرضى الذين يعانون من اضطرابات وراثية مختلفة منذ عام 2010، يقول الدكتور بونام مالك، مدير مركز الخلايا المنجلية في مستشفى سينسيناتي للأطفال، إن نهج تحرير الجينات كريسبر المستخدم في كاسجيفي يقتصر على اثنين إلى ثلاثة سنوات من الخبرة السريرية.
ومع ذلك، في هذه المرحلة، فإن Lyfgenia فقط هي التي تحمل تحذيرًا من الصندوق الأسود من إدارة الغذاء والدواء الأمريكية: نتيجة وفاة اثنين من المشاركين في نسخة سابقة من تجربة Lyfgenia السريرية بعد تشخيص إصابتهما بسرطان الدم. (اقترح صانعو الدواء أن هذه الوفيات لم تكن على الأرجح مرتبطة بالعلاج الجيني، ولكن ربما كانت مرتبطة بالمشاركين في العلاج الكيميائي اللازمين للتحضير له).
على الرغم من عدم الإبلاغ عن أي أحداث سلبية خطيرة خلال تجارب كاسجيفي السريرية، يقول ويليامز إنه من غير المعروف بعد ما إذا كان تحرير جينات كريسبر بشكل عام يحدث فقط في الأماكن الموجهة إليه، أو إذا حدث في مكان آخر من الجينوم أيضًا. إذا حدث هذا التحرير “خارج الهدف”، فمن المحتمل ألا يحدث بشكل متكرر، حيث لم تضع إدارة الغذاء والدواء الأمريكية (FDA) علامة عليه كمصدر قلق.
فتح الباب أمام علاجات كريسبر المستقبلية
إن حقيقة أن أول تطبيق تمت الموافقة عليه من قبل إدارة الغذاء والدواء لتحرير الجينات CRISPR هو علاج لـ SCD لم يكن المقصود منه أن يكون لفتة تحاول التعويض عن أكثر من قرن من تجاهل الحالة وعدد المرضى. ومع ذلك، يقول ويليامز: “إنه لأمر رائع أن يحدث ذلك”.
بعد حصول العلاج الأول باستخدام كريسبر على موافقة إدارة الغذاء والدواء الأمريكية، توقع ويليامز أن نظام تحرير الجينات “سوف يتم تطبيقه على أمراض أخرى بسرعة كبيرة”. وقد كان هذا هو الحال بالفعل؛ ولم يمض وقت طويل بعد ذلك، حتى وافقت إدارة الغذاء والدواء الأمريكية على استخدام علاج كريسبر لمرض ثانٍ: الثلاسيميا بيتا.
علاوة على ذلك، تشير الأبحاث الأولية إلى أن العلاجات التي تستخدم تحرير الجينات القائم على تقنية كريسبر قد يكون لديها القدرة على علاج أمراض وراثية أخرى تغير الحياة، مثل مرض الزهايمر، ومرض باركنسون، ومرض هنتنغتون، والتليف الكيسي.
وفي الوقت نفسه، يجب أن يكون التركيز على جعل عقاري كاسجيفي وليفجينيا – بالإضافة إلى الجيل التالي من العلاجات الجينية لمرض فقر الدم المنجلي – في متناول المرضى في جميع أنحاء العالم. يقول مالك إن هناك طرقًا يمكن من خلالها القيام بذلك، مثل جعل مجموعات الخلايا الجذعية أكثر كفاءة، وتقليل سمية التكييف قبل الزرع بحيث يمكن إعطاؤه في العيادات الخارجية.
فرغم أنه قد يكون من المغري أن ننظر إلى الحصول على موافقة إدارة الغذاء والدواء باعتباره لحظة “إنجاز المهمة”، فإن الحقيقة هي أنه خارج التجارب السريرية، فإن العلاجات الرائدة مثل كاسجيفي وليفجينيا لا يمكن أن تكون فعالة إلا بقدر ما يمكن الوصول إليها.
لمزيد من الأخبار والنشرات الإخبارية لـ CNN، قم بإنشاء حساب على CNN.com